NovaSeq X系列的首批产品在因美纳新一代高通量平台上引入了XLEAP-SBS化学技术。通过启用25B流动槽,NovaSeq X软件更新v1.2现在可以在双流动槽测序运行中产生多达16 Tb的数据。得益于此次更新,NovaSeq X Plus每年可以生成数万个全基因组。除了提高了通量,v1.2版本还升级了工作流程,从而提高了准确性并改进了Q值校准。通过结合提高的Q值准确性和XLEAP-SBS化学技术,NovaSeq X系列的“中碱基质量”和“高碱基质量”区间分别从Q20转变为Q24,从Q37转变为Q40。该仪器更高、更准确的碱基质量更好地反映了XLEAP-SBS化学技术和NovaSeq X系列平台的功能。
NextSeq 1000和NextSeq 2000基因测序仪上的控制软件v1.7可与XLEAP-SBS化学技术(将于2024年上半年推出)搭配使用,也具有相同的升级版工作流程,可提高准确性并改进Q值校准。
本文将重点介绍:
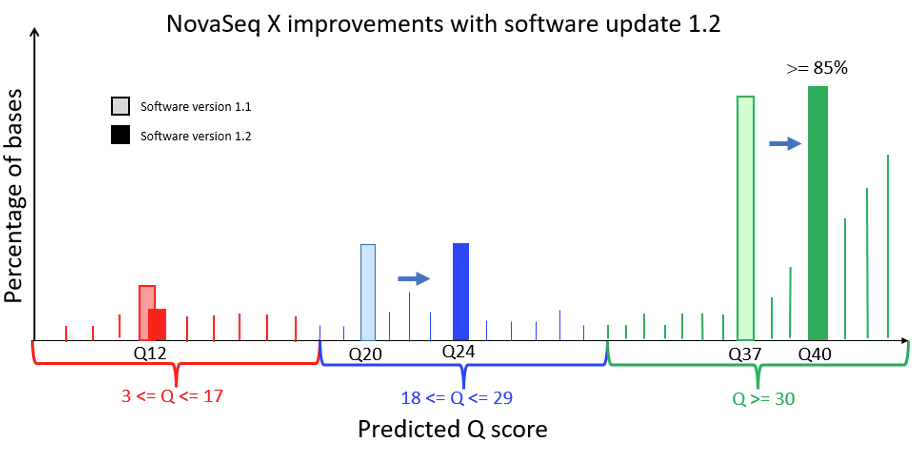
- 对测序和校准工作流程的改进使NovaSeq X系列能够在最高质量区间(Q40)中提供超过85%的碱基,并且与准确性的经验估计具有出色的相关性
- 在Q值校准过程中使用PCR-Free文库制备技术,以便Q值能更贴切地代表测序质量,而不是样本制备质量
- 将Q值量化为三个区间的数据管理模式和随之带来的成本效益
- Q值量化对下游应用的影响评估
- 测序和校准工作流程在NextSeq 1000和NextSeq 2000基因测序仪中的应用
下图1是Q值直方图,展示了NovaSeq X系列v1.2更新中的改进。本图也适用于NextSeq v1.5至v1.7控制软件更新改进。图中的细线表示质量分值达到特定整数值的碱基百分比。为了降低客户存储成本和数据传输时间,测序仪将这些整数间隔的质量分值聚合为三个区间,显示为深色条,标签分别为Q12、Q24和Q40。这些分区在概念上分别包含3至17、18至29和30及以上范围内的碱基质量。每个深色条下面的标签代表该聚合区间中所有碱基的质量分值。由于标签是区间数据的平均质量,因此区间会自然地聚合高于和低于标签值的各个高分辨率质量分值的碱基。由于Q值是对数尺度(详见下文),因此分布是不对称的,聚合分区大多由质量较高的碱基组成。v1.2版本更新使得碱基代表质量的评估更加准确,质量分值有所上升,目前最高质量的碱基报告为Q40。NovaSeq X系列能够产生>= 85%的Q40分值输出碱基;其中大部分碱基在高分辨率下超过Q40。
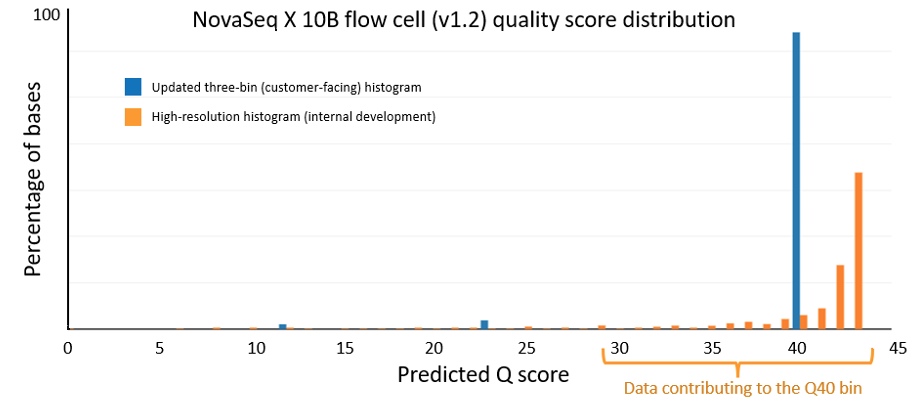
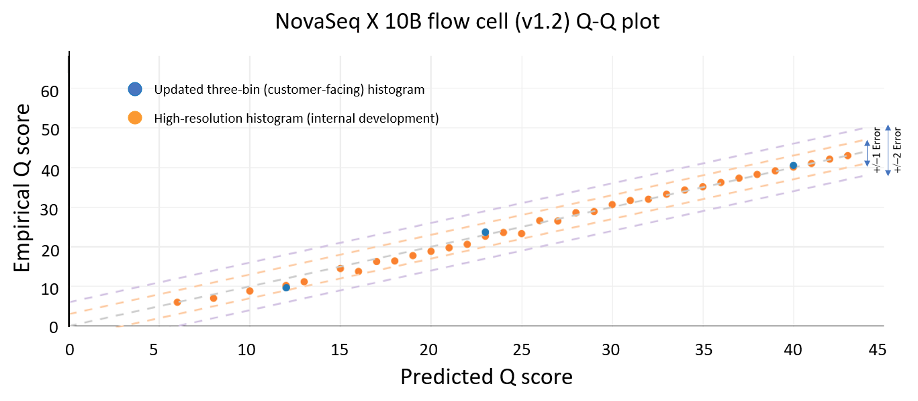
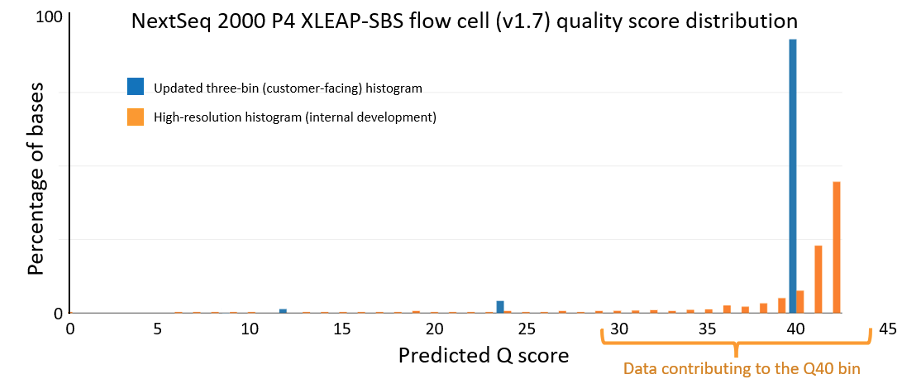
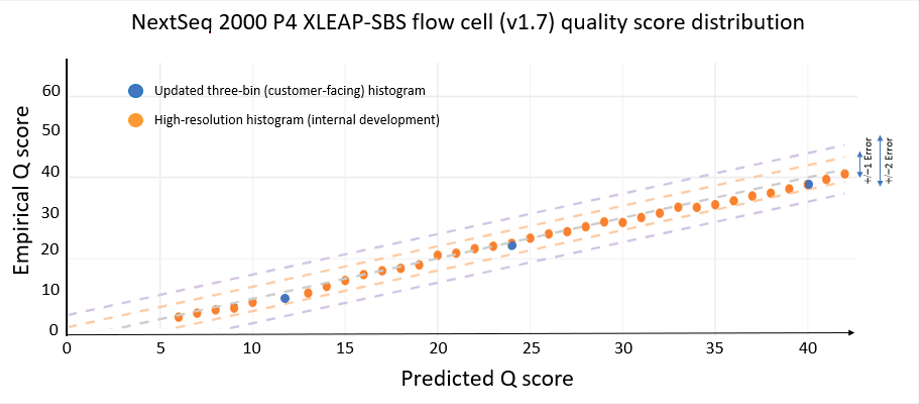
控制软件v1.7可在NextSeq 1000和NextSeq 2000基因测序仪上更新Q值
通过XLEAP-SBS化学技术和改进的校准方法实现的数据质量改进也能很好地应用于因美纳的旗舰款台式测序仪NextSeq 1000和NextSeq 2000。在NextSeq 2000上,从HG001样本制备的PCR-free文库可获得下图5所示的数据质量。使用P4 XLEAP-SBS流动槽验证了该结果,P3 XLEAP-SBS流动槽的性能与之相当。最高质量分区的平均Q值再次将经验错误率从Q34提高到Q40(万分之一的误差)。Q40区间中的碱基占运行输出碱基的90%以上。考虑到高分辨率直方图,这些碱基中的大多数实际上等于或超过Q40。
Q值基础知识:什么是碱基质量分值?什么是Phred分值?
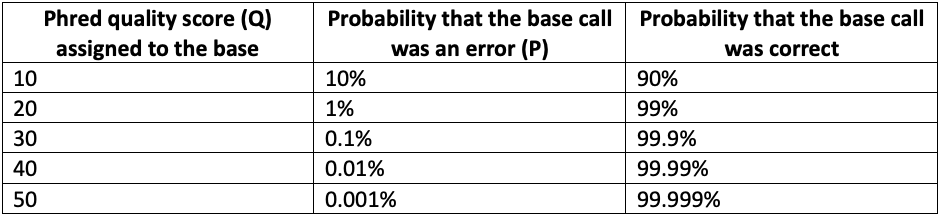
在因美纳测序仪上对每个碱基进行解码时,碱基检出模型会分配一个置信度分值,代表所分配的碱基检出正确的概率1。碱基检出错误的原始概率被转换成对数尺度,以压缩动态范围,就像功率测量中使用的dB命名法一样。由此得出的比例值称为碱基质量分值(或Q值,或Phred分值)。例如,当碱基检出程序对碱基检出正确性的置信度为99%时,出错的概率为1%,即Phred(Q)分值为20。
在数学上,Phred质量分值的定义是Q = -10 log10 P,其中P是碱基检出错误的概率。如下表1所示,随着Phred分值的增加,碱基检出的置信度也会以非线性方式增加。
Q值误差来源:并非所有误差都在测序仪上产生
经过正确校准的Q值模型可以预测测序误差的概率。不过,重要的是要明白,虽然有些误差是测序过程本身造成的——例如,噪音或信号功率低,但其他误差则源自测序仪的上游过程。NGS工作流程中常见的误差来源如下图7所示。
测序前,样本提取和文库制备将基因组DNA样本转化为片段文库。该过程通常涉及片段化、在每个片段的两端添加特定的接头以及扩增或纯化步骤。一些已发表的论文描述了这些过程可能带来的错误2-4。测序仪无法将样本制备过程中产生误差的碱基与样本中的其他碱基区分开来;样本制备误差是在上游悄然产生的。因此,必须在下游流程中通过纠错算法来估计和减少样本制备误差。

用于质量分值校准训练的方法自然包括样本制备和测序过程本身的误差(参见附录)。由于PCR-Free文库制备可将样本制备误差降到最低,因此仅使用PCR-Free数据进行校准得出的质量分值更能反映测序仪的实际碱基检出准确性。
下游分析中的重新校准过程可以动态地表征新数据集上的测序和样本制备误差,从而提高高度敏感应用的准确性。此外,这些校准过程可以针对特定的最终用户流程进行定制,修正样本制备误差特征的变化。Broad研究所的基因组分析工具包(GATK)最佳实践包括碱基质量得分重新校准(BQSR)步骤,该步骤可估算指定碱基质量与经验碱基质量之间的映射关系5。BQSR的难点在于它需要一个已知变异位点的数据库,而对于大多数样本来说,这个数据库并不容易获得。
DRAGEN流程包含先进的算法6,可以针对每个具体的流程/样本评估错误机制,无需参考真值集,并且尤其关注PCR相关的假象;DRAGEN既不需要也不使用BQSR。
Q值分辨率:大量基因组数据推动了碱基质量标记的优化,从而降低了存储需求
随着时间的推移,因美纳测序仪的数据通量大幅增加,因美纳采用了区间量化的质量分值,以此来减少客户的数据传输时间和存储成本。
早期的因美纳测序仪,如MiSeq基因测序仪,使用了50个整数区间生成Q值。为了大幅减少测序仪输出数据的存储空间占用,新仪器采用更少的区间来表示Q值的连续空间;这种方法降低了质量分值的熵,允许更高水平的压缩。NovaSeq 6000、NextSeq 1000、NextSeq 2000和iSeq均使用三区间Q值表。图8显示了将质量分值汇总到较少的区间数量中的优势。FASTQ文件(gzip格式)和BAM文件的存储大小减少了50%,这直接意味着存储成本的节省。
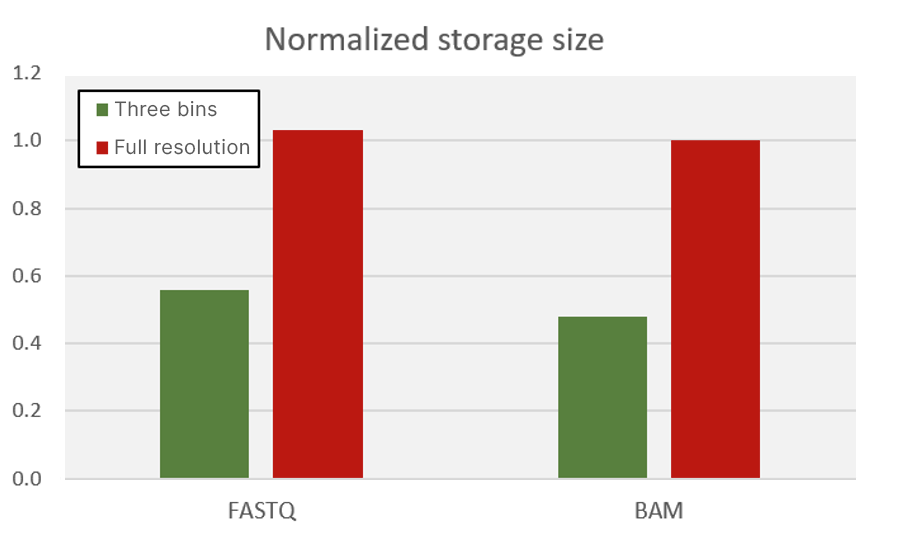
因美纳及其客户已经证明,Q值分辨率的降低对下游流程几乎没有影响,而且可以节省大量的数据传输时间(对大型测序项目和生产规模测序非常重要),同时还能大幅度减少所需的数据存储空间(对控制客户成本非常重要)。更多信息,请参阅因美纳有关此主题的应用说明7。
Q值在下游应用中的意义:Q值重要吗?
在使用最先进的变异检出程序的生殖系应用中,超过Q35的质量分值影响甚微。准确性主要受限于覆盖度,而不是碱基质量分值。覆盖度需求主要受泊松抽样的影响——例如,请参阅《计算生物学杂志》中的这篇论文8。随着灵敏度更高的体细胞应用(包括微小残留病)的发展(在这些应用中,变异的等位基因频率可能低得多),更高的Q值或许的确可以减少所需的原始测序数据量。针对这些新应用方向,DRAGEN体细胞测序流程也包含了一些算法,以提高对样本制备过程中引入的误差的检测能力9。
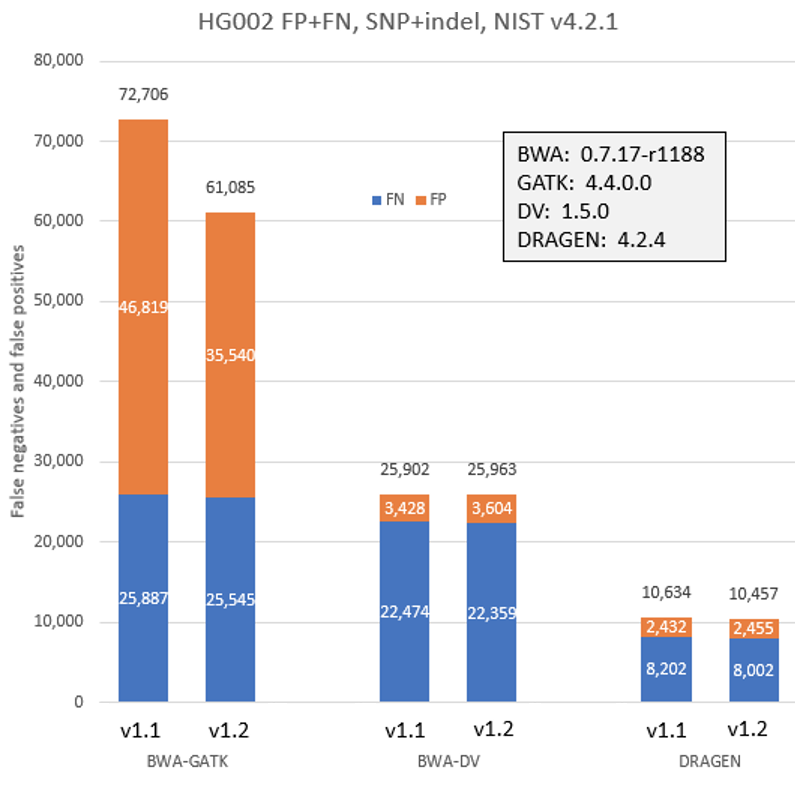
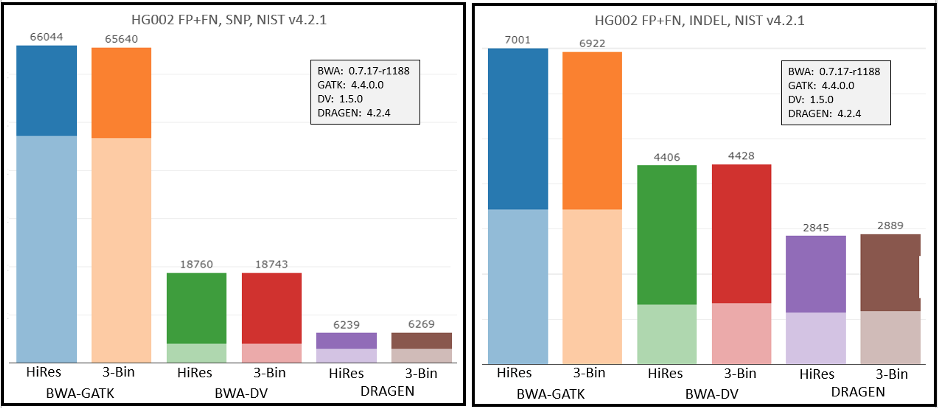
为了检验v1.2版本更新中的新工作流程和Q值预测改进对二级分析的影响,我们使用v1.1版和v1.2版的三种常用分析流程分析了HG002基因组样本。如下图9所示,随着Q值的提高,GATK的准确率有所提升。但DRAGEN和DeepVariant的性能在Q值提高后没有明显变化。
结论
NovaSeq X系列软件v1.2、NextSeq 1000和NextSeq 2000控制软件v1.7的更新更准确地反映了XLEAP-SBS化学技术的测序质量。凭借更准确的碱基检出、质量分值和分区,NovaSeq X、NextSeq 1000和NextSeq 2000基因测序仪现在可在最高质量(Q40)区间提供碱基。
此外,NovaSeq X系列软件v1.2和NextSeq 1000和2000控制软件v1.7在分配质量分值时纳入了以下重要考虑因素:
- 碱基质量分值是在测序仪上计算的基于统计测序错误率的一种评估
- 为了最大限度地减少样本制备过程中引入的误差,因美纳的校准过程中采用了PCR-Free文库制备方法
- 高效的Q值分区对于降低客户存储成本和减少数据传输时间非常重要
- 分区对使用最先进的变异检出程序的生殖系应用影响很小或没有影响
有关NovaSeq X系列软件v1.2更新的更多信息,请点击此处查阅。
附录:Q值校准训练
在向客户发布之前,测序仪平台会通过校准训练来优化碱基质量分值预测。这可确保测序平台上分配的碱基质量分值与经验碱基检出准确度密切吻合。
许多碱基质量分值校准训练算法都源自1998年发表的一篇基础论文,该论文描述了一种高效的计算质量分值的分配方法10。它使用少量预测变量(如信噪比)来构建一个查找表。每一行都记录了预测空间中一小块区域的经验质量。它为给定一组量化预测值生成碱基质量分值提供了一种简便的方法。虽然该论文仅使用了从色谱文件中提取的四个启发式预测变量,但这种通用方法可以在任何类型的测序仪中使用任意数量的预测变量。
经验质量的定义是,在给定的预测变量空间区域内,被正确检出的碱基数与被检出的碱基总数之比。用于计算的碱基仅限于已知真值的碱基,例如,用于人类数据的Genome in a Bottle(GIAB)真值集,它涵盖了人类基因组的大部分内容。具有自己的高质量真值集的其他基因组(细菌、微生物)也包含在其中,以确保k-mers在训练中得到尽可能公平的体现。容易出现映射错误的区域将从计算中剔除,因为映射不是测序操作的一部分,不应影响测序错误率的测量。
历史版本:
2024年1月11日:本文标题已从“NovaSeq X v1.2软件可实现超过80%的碱基质量>= Q40的测序结果”更新为当前标题。
2023年12月21日:本文更新了有关NextSeq 1000基因测序仪和NextSeq 2000基因测序仪的控制软件v1.5和v1.7版本的信息。
参考文献:
- Illumina. Understanding Illumina Quality Scores. illumina.com/content/dam/illumina-marketing/documents/products/technotes/technote_understanding_quality_scores.pdf. Published 2014. Accessed October 9, 2023.
- Chen G, Mosier S, Gocke C, Lin M-T, Eshleman J. Cytosine Deamination is a Major Cause of Baseline Noise in Next-Generation Sequencing. Mol Diagn Ther. 2014 Oct; 18(5): 587–593. doi:10.1007/s40291-014-0115-2
- Otsubo Y, Matsumura S, Ikeda N, Yamane M. Single-strand specific nuclease enhances accuracy of error-corrected sequencing and improves rare mutation-detection sensitivity. Arch Toxicol. 96, 377–386 (2022). doi:10.1007/s00204-021-03185-y
- Gregory T, Ngankeu A, Orwick S, et al. Characterization and mitigation of fragmentation enxyme-induced dual stranded artifacts. NAR Genom Bioinform. 2020 Dec; 2(4). doi: 10.1093/nargab/lqaa070
- Caetano-Anolles D. Base Quality Score Recalibration (BQSR) – GATK. Broad Institute Genome Analysis Toolkit. gatk.broadinstitute.org/hc/en-us/articles/360035890531-Base-Quality-Score-Recalibration-BQSR-. Published 2023. Accessed October 9, 2023.
- https://science-docs.illumina.com/documents/Informatics/dragen-v3-accuracy-appnote-html-970-2019-006/Content/Source/Informatics/Dragen/dragen-v3-accuracy-appnote-970-2019-006/dragen-v3-accuracy-appnote-970-2019-006.html
- Illumina. NovaSeq™ 6000 System Quality Scores and RTA3 Software. illumina.com/content/dam/illumina-marketing/documents/products/appnotes/novaseq-hiseq-q30-app-note-770-2017-010.pdf. Published 2017. Accessed October 9, 2023.
- Deng C, Daley T, Calabrese P, Ren J, Smith A. Predicting the Number of Bases to Attain Sufficient Coverage in High-Throughput Sequencing Experiments. J Comput Biol. 2020 Jul; 27(7): 1130–1143. doi: 10.1089/cmb.2019.0264
- Scheffler K, Catreux S, O’Connell T, et al. Somatic small-variant calling methods in Illumina DRAGEN™ Secondary Analysis. bioRXiv 2023.03.23.534011. doi:10.1101/2023.03.23.534011
- Ewing B, Hillier L, Wendl M, Green P. Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Res. 1998;8:175–187. doi:10.1101/gr.8.3.175