Best practices to minimize rRNA contamination in TruSeq Stranded Total RNA libraries
11/10/21
In the TruSeq Stranded Total RNA library preparation workflow, the first step is to deplete rRNA from the total RNA input using Ribo-Zero. Sometimes, higher levels of rRNA are found in the final sequencing data. This bulletin addresses how to determine the root cause and best practices to avoid rRNA contamination.
rRNA depletion with Ribo-Zero:

Figure 1. a) Biotinylated Ribo-Zero probes are mixed with Total RNA and hybridize to the complementary target rRNA regions. b) Streptavidin-coated magnetic beads are added to the solution of rRNA-RiboZero Probes to capture the Biotinylated probes. c), d) The magnetic beads-probes-rRNA complexes are removed leaving the rRNA depleted RNA in the supernatant. rRNA depletion is not 100% efficient; a low level of rRNA will remain in the supernatant.
Potential root causes of rRNA reads in excess:
- Binding of the endogenous rRNA by the rRNA removal probes is not optimal.
- Binding of the rRNA removal probes by the magnetic beads is not optimal.
- Magnetic beads were not completely removed from the sample.
- DNA contamination
The source of rRNA reads in sequencing data assists with troubleshooting:
| Source of rRNA reads in sequencing data | ||
|---|---|---|
| Source | Alignment in Read 1 | Alignment in Read 2 |
| Endogenous rRNA | Anti-sense | Sense |
| rRNA removal probes | Sense | Anti-sense |
- Binding of the endogenous rRNA by the rRNA removal probes is not optimal
If the conditions at which the probes bind the endogenous rRNA are suboptimal, or the complementarity to the target rRNA is low, depletion will be inefficient. While probes will be removed, unbound endogenous rRNA will remain in solution.
Because the TruSeq Stranded Total RNA kits are strand-specific, the strandedness of rRNA reads will match the endogenous rRNA orientation: Read 1 will map to the antisense strand and Read 2 will match to the sense strand.
How to avoid this issue:
- Mix the reagents completely to allow the probes to contact the rRNAs
- Use the correct input amount (probes are optimized for a specific amount of total RNA)
- Set the correct temperature on heat block or thermal cycler to allow optimal probe binding
- Use rRNA removal solution that is not expired and has been stored properly
- Ensure that species is compatible with rRNA removal probes
- Binding of the rRNA removal probes by the magnetic beads is not optimal
If the beads do not capture the probes efficiently, the sample will have higher residual levels of both endogenous rRNA and complementary rRNA probes that will be incorporated into the final libraries.
Read strandedness in this situation is mixed depending on the amount of probes left in the sample, and Read 1 and Read 2 will align to both strands because both endogenous rRNA and the complementary rRNA removal probes are sequenced.
How to avoid this issue:
- Follow best practices for bead handling
- keep beads at room temperature for at least 30 minutes
- mix beads very well before use
- mix beads and samples completely
- Store beads according to recommendations in the pertinent library preparation reference guide
- Do not use rRNA removal beads that have been frozen
- Use the validated magnetic stand to capture beads (magnetic stand information can be found in the Consumables and Equipment list in the reference guide)
- Follow best practices for bead handling
- Magnetic Beads were not completely removed from the sample
When beads are not properly captured by the magnet, they are visualized in traces (from the TapeStation, Bioanalyzer, Fragment Analyzer, or comparable instrument) as a broad peak at high molecular weight size (see trace below). Read strandedness is mixed depending on the amount of probes left in the sample, and Read 1 and Read 2 will map to both strands because both endogenous rRNA and the complementary rRNA removal probes are sequenced.
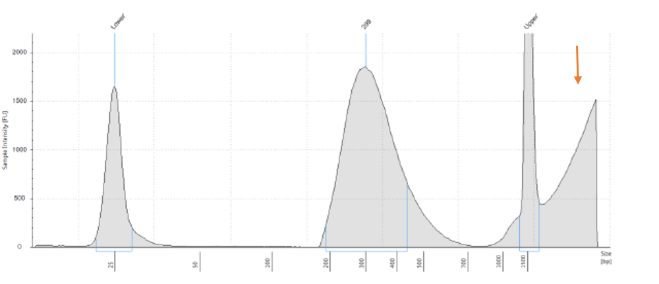
How to avoid this issue:
- Use the validated magnetic stand to capture beads (magnetic stand information can be found in the Consumables and Equipment list in the reference guide)
- Visually inspect supernatant after removing from the rRNA removal beads to ensure that no rRNA removal beads were carried into the supernatant
- DNA contamination
If there is DNA present in the RNA input, libraries are generated from the contaminating DNA. Reads from library fragments generated from DNA show mixed strandedness. In this case, changes in strand specificity are observed not only for rRNA but also for transcripts (exons). Additionally, regions corresponding to intronic or intergenic regions may be overrepresented in the data.
How to avoid this issue:
- Perform DNase treatment on input RNA before library preparation

Please refer to the following troubleshooting diagram:
