癌症表观遗传学
癌症样本的表观遗传学分析
癌症表观遗传学变化的研究(如异常甲基化和转录因子结合改变)可以提供对重要致癌通路的了解。甲基化变化常常会使基因激活或沉默,表观基因组的变化可以影响基因表达和癌症进展的速度。
新一代测序(NGS)和芯片技术可以检测到甲基化模式和癌症中其他表观遗传学的变化。Illumina与癌症表观遗传学专家进行了合作,确保其NGS和芯片解决方案能满足该领域快速变化的需求。
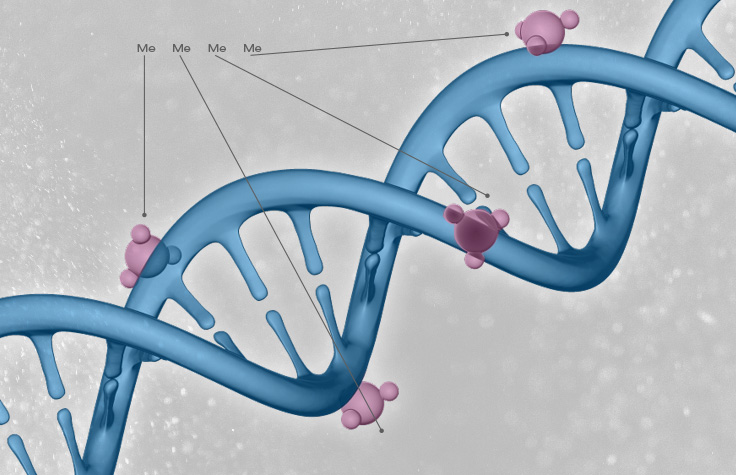
追踪肿瘤进展
Christopher Mason博士介绍了如何通过转录组、表观基因组和表观转录组测序来追踪肿瘤进展。


检测肿瘤中DNA-蛋白质相互作用的变化
转录因子结合的改变是另一个与癌症相关的常见表观遗传学变化。染色质免疫沉淀测序(ChIP-Seq)能提供癌症细胞和正常细胞在整个基因组内与DNA相关的蛋白活性的信息。
这种方法能让研究人员以一种无假设的方法来研究基因表达调控。深度测序能检测通常在转录因子中观察到的低丰度蛋白质-DNA相互作用。
深入了解ChIP-Seq对癌症表观基因组进行测序
在第49集Illumina基因组学播客中,Susan Clark博士介绍了表观遗传学在癌症和人类生物学领域的作用。
癌症表观基因组学和基因表达中的“杂音”
在Illumina基因组学播客的第21集节目中,我们与来自俄亥俄州克利夫兰凯斯西储大学的遗传学与基因组科学教授Peter Scacheri博士探讨了癌症的表观遗传学和表观基因组学。
立即收听
分析可及性染色质
染色质转座酶可及性测序分析(ATAC-Seq)涉及对整个基因组的开放染色质区域进行测序。这些信息可以帮助研究人员了解染色质包装如何影响癌症基因表达的见解。这种方法不需要事先了解调控元件,因此是一种强大的表观遗传学发现工具。
深入了解ATAC-Seq
甲基化芯片在脑肿瘤研究中的应用
来自海德堡大学的Andreas von Deimling教授展示了在神经肿瘤学研究中使用甲基化芯片和NGS获得的结果。对于未来如何将这些分析应用于脑肿瘤诊断,他分享了自己的观点。
观看视频