Introduction
Sequences consisting of repetitions of relatively short pieces of DNA, known as tandem repeats (TRs), occur throughout the genome (eg, Figure 1). TR mutation rates can be 10’s to 1000’s times higher than other genomic regions making TRs large contributors1 to the human genetic variation. TRs largely mutate through “slippage” where the number of repeats increases or decreases between generations. Accumulating evidence shows that TRs play a role in basic cellular processes2,3 and large expansions of tandem repeats are linked to a variety of neurological disorders including amyotrophic lateral sclerosis (ALS), fragile X syndrome, and various forms of ataxia.
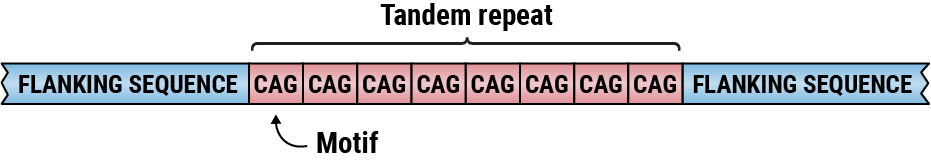
A tandem repeat with CAG motif.
Sequencing a region containing a TR produces a collection of reads that either partially or completely overlap the repeat sequence (Figure 2). By piecing together alignments of these reads we can determine the length of the repeat on each haplotype. Our group has developed several methods for both targeted4,5 and genome-wide6 TR analysis. Here we focus on ExpansionHunter 4,5, a method for targeted analysis of regions containing one or multiple adjacent TRs that can estimate sizes of repeats both shorter and longer than the read length.
TR genotyping is a very difficult problem and even the best methods can occasionally make incorrect genotype calls. Because of this, it is important to have robust visualization methods for inspecting alignments of the reads used to genotype the repeat in question. Additionally, such visualization methods make it possible to detect changes in the repeat motif (eg, interruptions) which can have clinically significant effects7,8. The standard data visualization pipelines are usually limited to displaying alignments of reads to the reference genome and thus are inadequate for repeats expanded relative to the reference or repeats with alleles of different lengths. To address these issues, we have developed the Repeat Expansion Viewer (REViewer) -- a tool for visualizing the graph realigned reads output by ExpansionHunter. REViewer determines haplotype sequences by phasing adjacent repeats and then distributes read alignments to these haplotypes. The resulting static images make it possible to visually assess the accuracy of a given genotype call and to identify if the repeat sequence contains any interruptions.
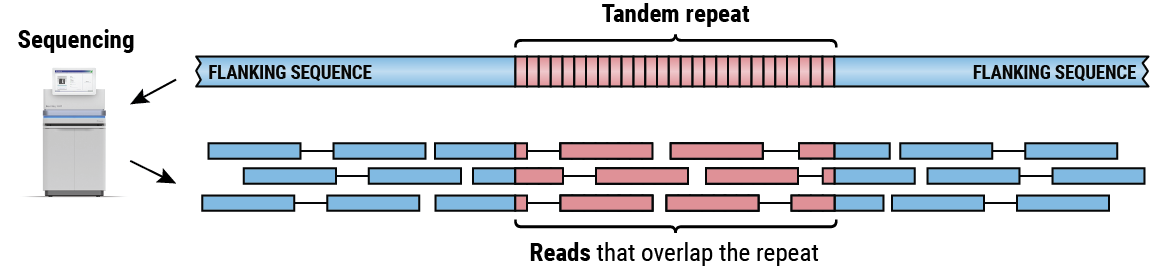
Paired reads generated by sequencing a tandem repeat that is longer than the read length.
Visualizing alignments of reads in tandem repeat regions
REViewer is designed to display alignments of reads generated by ExpansionHunter (Figure 3, boxes 1-3). These alignments are obtained by realigning reads originating in the target region to the corresponding sequence graph encoding one or more repeats located there5. REViewer then constructs putative haplotype sequences using repeat genotypes produced by ExpansionHunter and then selects a pair of haplotypes that have the highest consistency with the read alignments (Figure 3, boxes 4-6). (This step is skipped for repeats on haploid chromosomes.) Next, REViewer determines the set of possible alignment positions for each read pair on each haplotype. For example, a read pair originating within a flanking sequence shared by both haplotypes has exactly one alignment position on each haplotype (Figure 3, Box 7a) while a read pair whose both mates are comprised of the repeat sequence has multiple possible origins on haplotypes with sufficiently long repeats (Figure 3, Box 7b). To generate a read pileup, REViewer selects one alignment position at random for each read pair. This step is repeated a specified number of times (10,000 by default) to generate multiple pileups. The pileup with the most even coverage of each haplotype is selected for visualization (Figure 3, Box 8).
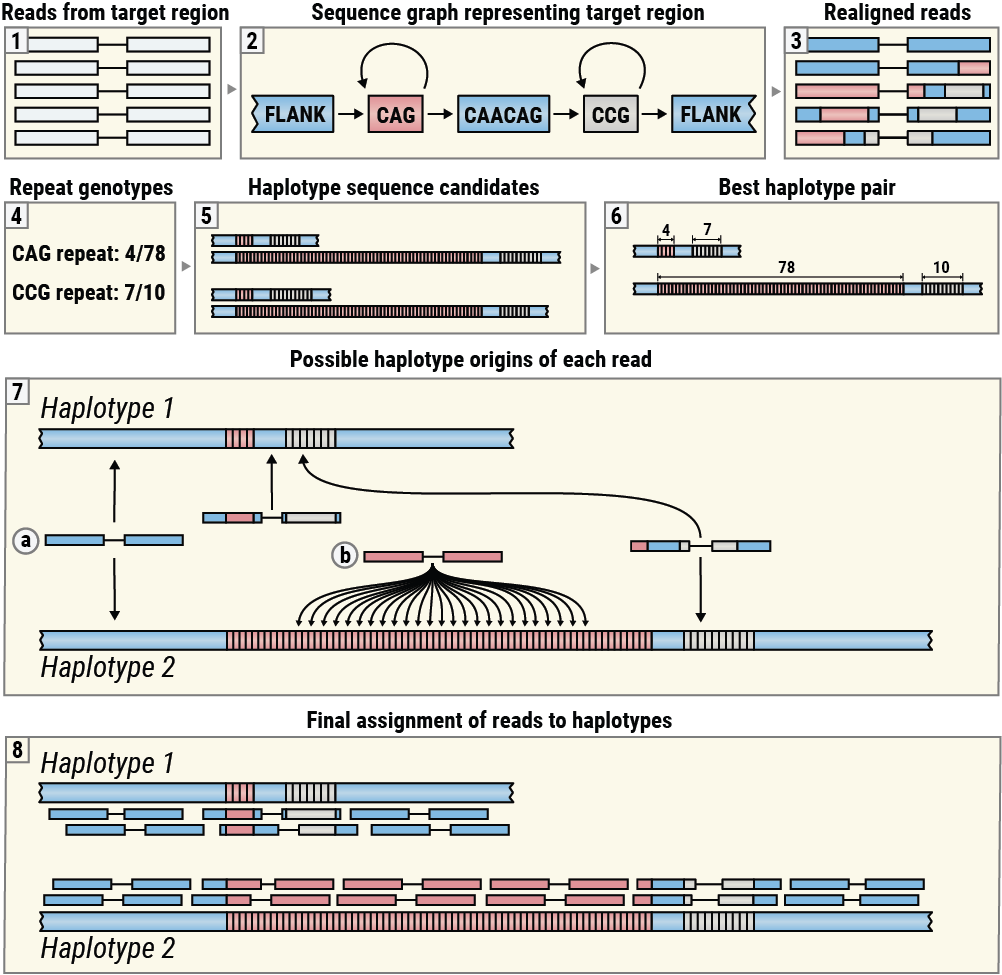
An overview of the REViewer visualization method.
This algorithm is based on the idea that if a given locus is sequenced well and each constituent repeat is genotyped correctly, then it is possible to distribute the reads to achieve an even coverage of each haplotype. (Although many reads may not be assigned to the correct haplotype of origin, especially in cases when the repeats are homozygous, and the resulting haplotypes are identical.) Conversely, if the size of a repeat is significantly overestimated or underestimated, no assignment of reads will result in an even pileup making the genotyping error easy to notice.
Visualization of accurately genotyped repeats
For the remainder of the article, we will review examples of read pileups generated by REViewer from real4 and simulated data. We start with pileups corresponding to accurate genotypes that are well supported by the reads.
A short repeat
Consider a read pileup for ATXN3 repeat whose alleles are shorter than the read length depicted on Figure 4. This repeat is genotyped 20/20 (20 motif copies on each allele). Each panel of this plot corresponds to a haplotype (the haplotype sequence shown in the top row). The haplotype sequences and the reads are colored according to their overlap with the repeat (orange) or the surrounding flanking sequence (blue). All mismatching bases in reads are shown.
The pileup plot shows that the genotype call is well supported by the reads because each allele is supported by many spanning reads (reads that span the repeat in its entirety) and because there are no reads with discrepant alignments. (A discrepant alignment means that the read is inconsistent with either of the two haplotypes – e.g., a read with 40 repeats would be inconsistent with the genotype 20/20.) There is clear evidence of interruptions in the repeat sequence. For example, the cytosine in the third to last motif is mutated into a thymine.
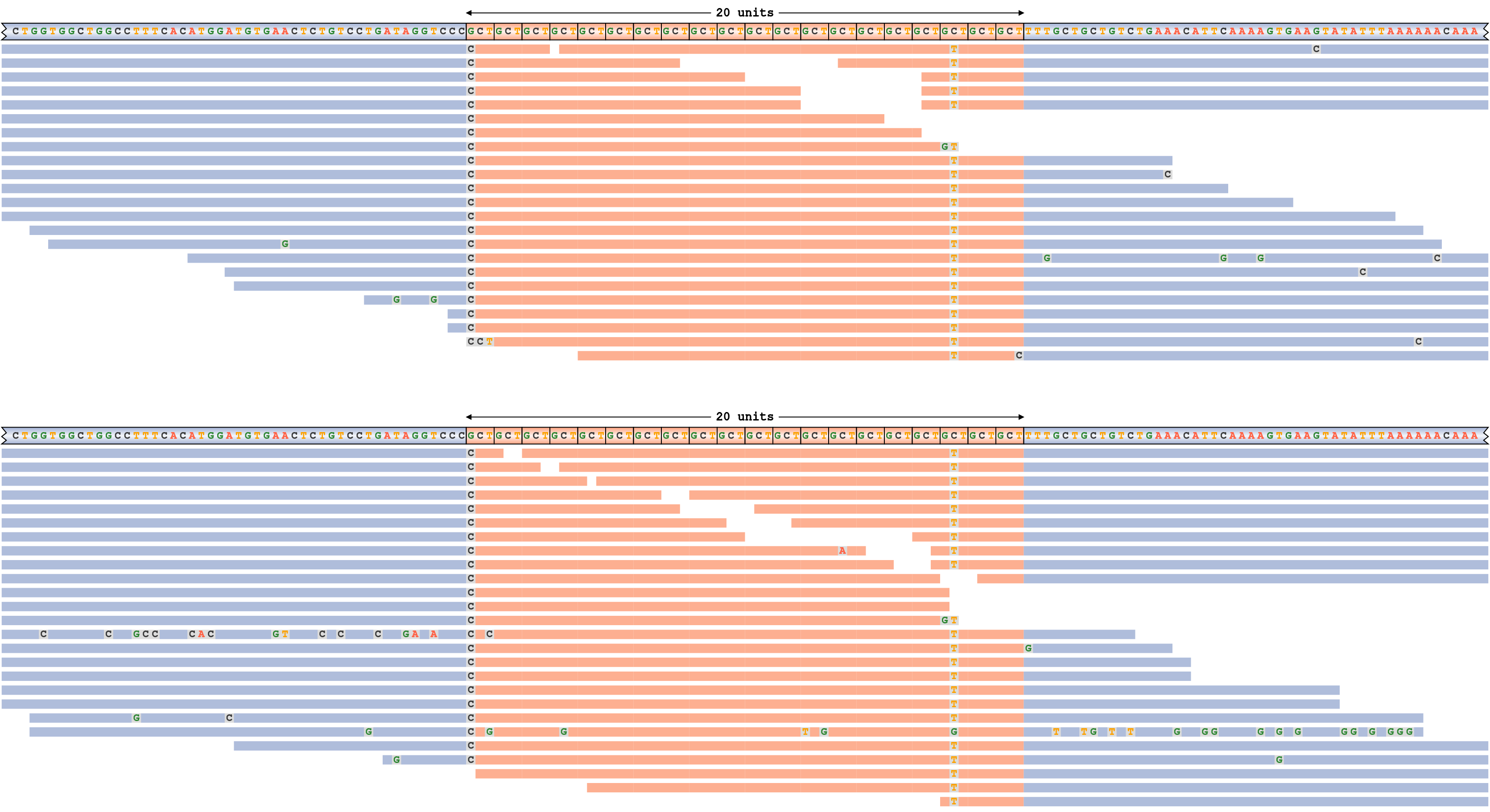
A read pileup for ATXN3 repeat with genotype 20/20. The sequence interruptions correspond to positions with mismatches in most of the read alignments.
An expanded repeat
Figure 5 depicts DMPK repeat with an expanded allele. The expanded repeat is well supported by the reads because REViewer was able to distribute the reads throughout the repeat to achieve similar read coverage across the entire haplotype. (It is important to remember that the alignment positions of reads within the repeat are chosen randomly.) The short allele is also well supported by a large number of spanning reads. Alignments depicted in fainter colors correspond to reads that could be assigned to either allele.
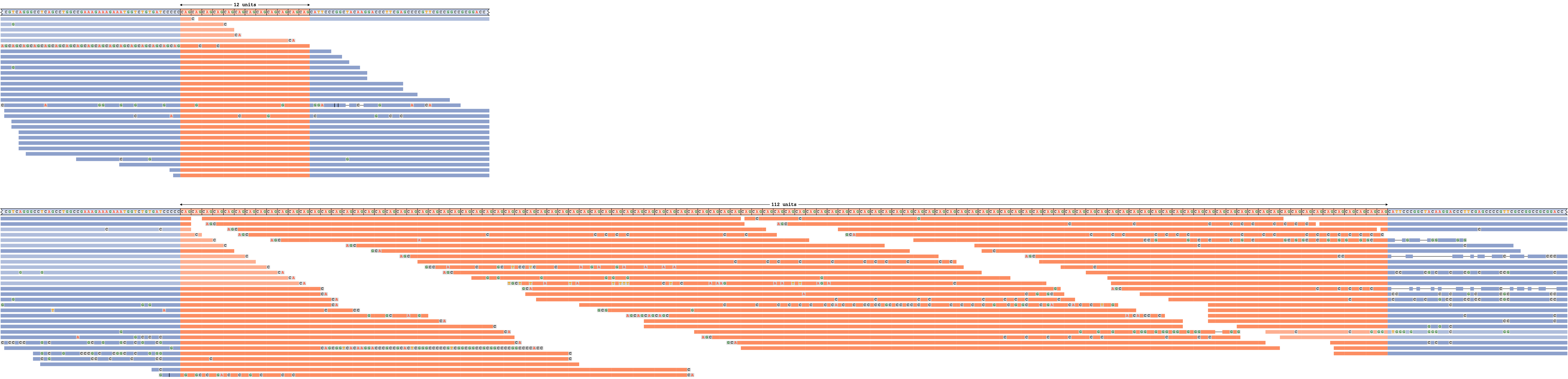
A read pileup for an DMPK repeat with an expansion on one allele.
A locus with two adjacent repeats
To demonstrate a more complex application of REViewer, we applied it to the HTT repeat region containing two adjacent repeats: the pathogenic CAG repeat and the nearby “nuisance” CCG repeat. The former repeat is genotyped as 14/17 and the latter repeat is genotyped as 9/12. Consequently, one of the haplotypes shown on Figure 6 contains repeats of size 14 and 12 respectively while the other haplotype contains repeats of size 17 and 9. It is evident that both haplotypes are well supported by the reads. Additionally, the pileup plot shows that there is a G to A mutation in the second copy of the CCG repeat motif on both haplotypes.
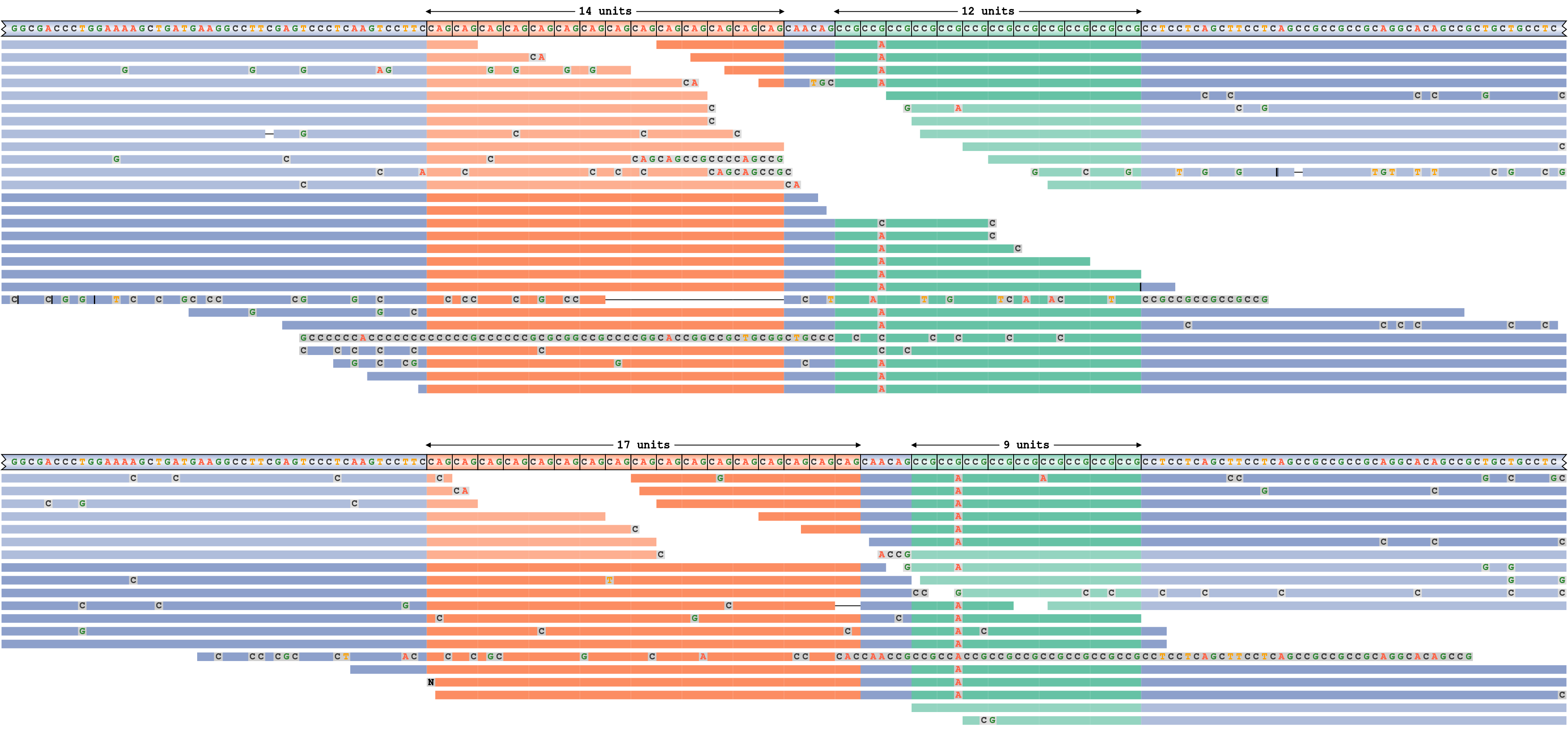
A read pileup for HTT locus containing two nearby repeats.
Visualization of inaccurately genotyped repeats
This section describes examples of read pileups corresponding to inaccurately genotyped repeats. We use simulated data to illustrate both false positive and false negative repeat expansion calls.
An overestimated repeat size
To give an example of a pileup corresponding to a false-positive repeat expansion call, we simulated reads from the C9ORF72 repeat region with homozygous genotype 10/10. We then spiked in a nearly perfect C homopolymer read that has a somewhat close resemblance to the C9ORF72 repeat sequence and ran REViewer forcing the repeat genotype to be 10/30 instead of 10/10. Figure 7 depicts the corresponding read pileup. As expected, the pileup shows that all but one of the reads placed on the haplotype with the longer repeat are also consistent with the shorter haplotype (these reads are depicted in fainter colors) and that only one poorly aligned read supports the expansion. In practice this would be considered a likely false positive call caused by a single low quality read.
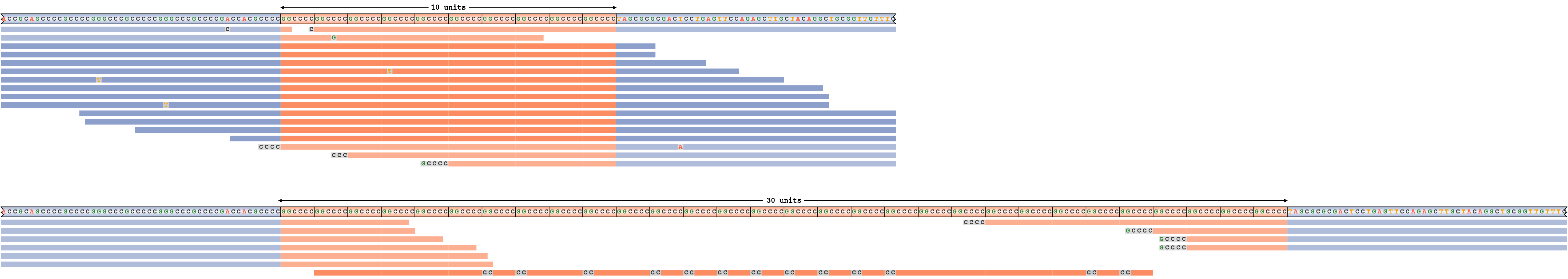
Incorrectly called expansion of C9ORF72 repeat.
An underestimated repeat size
To generate an example of a false-negative repeat expansion call, we simulated an FMR1 repeat with genotype 15/55 and then forced REViewer to generate a read pileup corresponding to an (incorrect) genotype 15/30. Figure 8 shows the resulting pileup. Notice that in order to reconcile the reads originating within the repeat of size 55, REViewer clipped the ends of alignments to the size of the longest allele. The actual clipped parts of the reads are displayed as gray segments with base sequences shown. Because there is an excess of reads overlapping the repeat with 30 motifs and because all these reads consist of the repeat sequence, we conclude that the size of the repeat is likely to be underestimated.
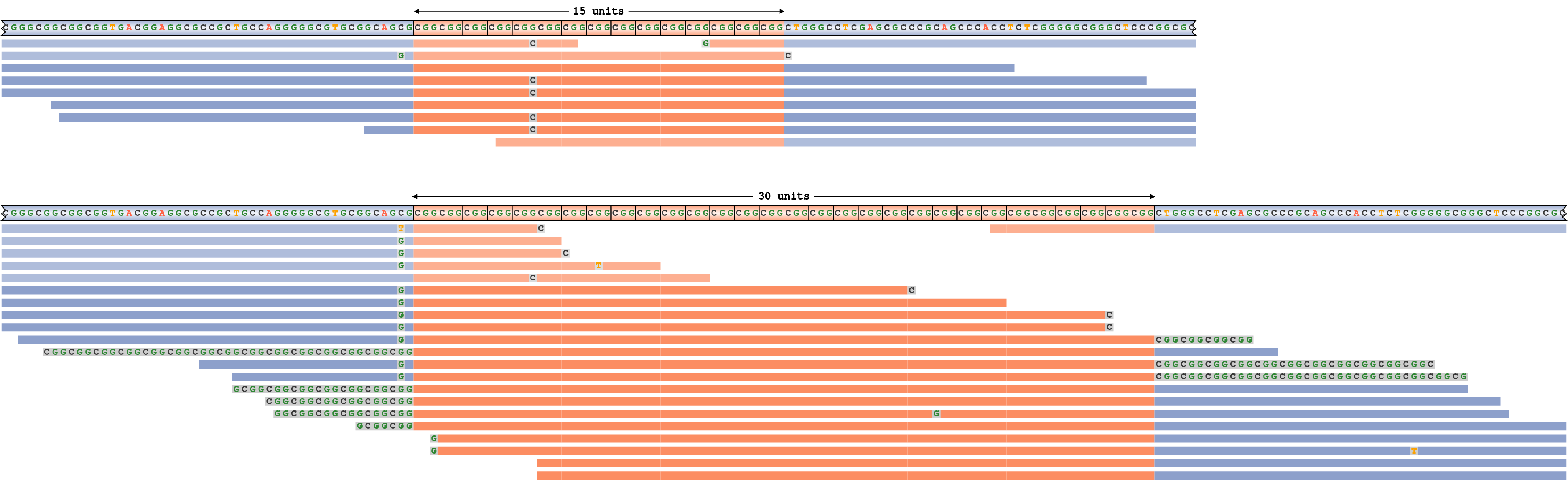
An FMR1 repeat pileup corresponding to the genotype where the size of the longest allele is underestimated.
Limitations
REViewer is a tool for assessing consistency of sequencing data with repeat genotypes produced by ExpansionHunter. It provides a mechanism for reviewing the evidence supporting a genotype call in clinical settings and identifying problematic corner cases to drive future development. The read pileup plots generated by REViewer may contain inaccuracies: The repeats may not be phased correctly (eg, when repeats are located far apart from each other) and read pairs consistent with both haplotypes will often be assigned to the incorrect haplotype. Also, the current version of REViewer visualizes repeats whose span does not exceed the fragment length (longer repeats are capped at the fragment length).
Conclusions
We developed a tool for visualizing alignments of reads supporting repeat genotypes determined by ExpansionHunter. In order to display full-length alignments, REViewer determines haplotype sequences of the target locus by phasing genotypes of all repeats located in close proximity to each other. REViewer distributes reads between haplotypes while randomly choosing locations of reads with multiple possible origins so that the reads are spread out as evenly as possible both between and within haplotypes. While the placement of many individual reads may be incorrect, the plot makes it possible to perform efficient visual assessment of a given genotype call. We also demonstrated that it is possible to use these plots for detecting interruptions in the repeat sequence and in the sequence immediately surrounding the repeat. Work is underway to develop and validate the ability to call interrupting sequences where their presence may have clinical implications.
Finally, we’d like to note that REViewer can be used to visualize short indels (because ExpansionHunter has rudimentary support for this variant type) and can be made to work with other variant types in principle. If you encounter any issues with using REViewer or have suggestions for improving the program, don’t hesitate to reach out to us (Egor edolzhenko@illumina.com and Mike meberle@illumina.com).
External links
- ExpansionHunter: https://github.com/Illumina/ExpansionHunter
- REViewer: https://github.com/Illumina/REViewer
References
- Fan H, Chu JY. A brief review of short tandem repeat mutation. Genomics Proteomics Bioinformatics. 2007 Feb;5(1):7-14. doi: 10.1016/S1672-0229(07)60009-6.
- Gymrek M, Willems T, Guilmatre A, et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat Genet. 2016 Jan;48(1):22-9.
- Hannan AJ. Tandem repeats mediating genetic plasticity in health and disease. Nat Rev Genet.2018 May;19(5):286-298.
- Dolzhenko E, van Vugt JJFA, Shaw RJ, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017 Nov;27(11):1895-1903.
- Dolzhenko E, Deshpande V, Schlesinger F, et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics. 2019 Nov 1;35(22):4754-4756.
- Dolzhenko E, Bennett MF, Richmond PA, et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 2020 Apr 28;21(1):102.
- Kraus-Perrotta C, Lagalwar S. Expansion, mosaicism and interruption: mechanisms of the CAG repeat mutation in spinocerebellar ataxia type 1. Cerebellum Ataxias. 2016 Nov 22;3:20.
- Wright GEB, Collins JA, Kay C, et al. Length of Uninterrupted CAG, Independent of Polyglutamine Size, Results in Increased Somatic Instability, Hastening Onset of Huntington Disease. Am J Hum Genet. 2019 Jun 6;104(6):1116-1126.