How short inserts affect sequencing performance
12/18/20
Sequencing libraries typically contain a mixture of different-sized library fragments. Shorter library fragments cluster more efficiently than longer libraries, and a high proportion of short libraries in the final library pool can negatively affect overall run metrics. If the sequencing read length is longer than the library insert size, sequencing can continue through the full inserts, read the adapter sequence on the other side of the insert, and may run into the flow cell. When sequencing continues into the flow cell, the read runs out of template for incorporation of bases, causing an intensity drop and potential loss of signal registration. Run results can show a sharp decline in the Q30 metric, which can be accompanied by focusing errors and possibly cause the run to abort. Before setting up the sequencing run, check to make sure that correct read length and run parameters are used.
- Library considerations for sequencing
It is important to consider both the library insert size and the desired sequencing read length before library preparation. Each of these factors will affect the quality of the sequencing run and the data output.
Libraries prepared for sequencing consist of DNA inserts and ~60–75 bp of adapter sequences flanking the insert on each end (approximately 120–150 bp total, Figure 1A). These adapters include the p5 and p7 sequences required to bind to the flow cell, the unique index or indexes, and the sequencing primer binding sites.

Figure 1. Adapter ligation during library preparation. The adapters are added to the DNA insert during library preparation. A. The DNA insert is prepared by adding an A-tail and phosphorylation. B. The adapter complex which includes the P5/P7 flow cell binding adapter is added to the DNA insert. C. The DNA insert is ready for sequencing. D. The DNA insert binds to the flow cell for sequencing. Primers bind to the DNA insert to generate reads.
- Sequencing read length and short fragment binding efficiency
The most appropriate sequencing read length depends on the shortest insert length, the average insert length, the read length requirements for the analysis method, and the application. Read length recommendations are available for all Illumina library preparation methods.
- How to choose sequencing run read length
Perform a quality check of the final libraries to assess the sizing profile. The final sizing profile represents the DNA insert length plus the length of the adapter sequences. During sequencing, Read 1 starts at the beginning of the insert sequence (Figure 1B). The most appropriate sequencing read length to use depends on the shortest insert length, with the selected read length being shorter than the average DNA insert.
- Short Fragments bind more efficiently on the flow cell
Short fragments preferentially bind to the flow cell compared to larger fragments. This negatively affects run performance as shorter fragments are overrepresented on the flow cell and in the final sequencing data. If DNA inserts are shorter than the run read length, sequencing continues through the DNA insert, proceeds through the adapter sequence, and can run into the flow cell. If libraries contain a high percentage of short fragments that are not critical for an experiment, shorter sized fragments can be removed from the pool before sequencing.
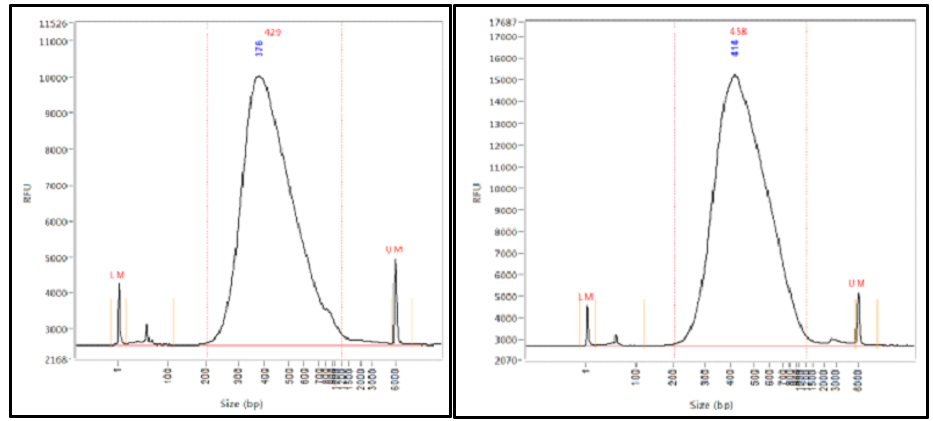
Figure 2. Library distribution. In the left and right panels, the library distributions start at 200 bp and extend to 800 bp. The average sizes are 429 bp and 458 bp, respectively. Because adapters contribute ~150 bp to the final library size, the insert lengths are 279 bp and 308 bp, respectively. A 2x250 bp run set-up is the longest read length recommended for these libraries allowing for some overlap.
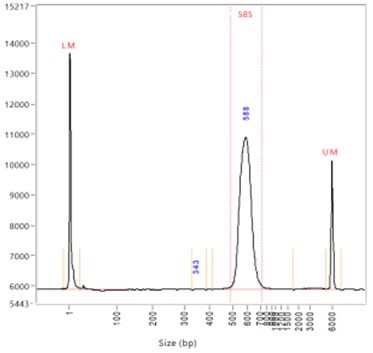
Figure 3. Narrow library distribution. This library has an average size of 585 bp and the insert length is 435 bp. This library can be sequenced with a 2x300bp run. Paired 300 bp reads sequence the full insert length without reading into the adapter sequences.
- How to choose sequencing run read length
- Adapter dimers
A peak at around 120–150 bp indicates the presence of adapter dimers (Figure 4). Adapter dimers are short fragments that form when two adapters ligate to each other without an insert. Removal of adapter dimers before sequencing is recommended. For more information on adapter dimers, refer to the bulletin: Adapter dimers: causes, effects, and how to remove them.
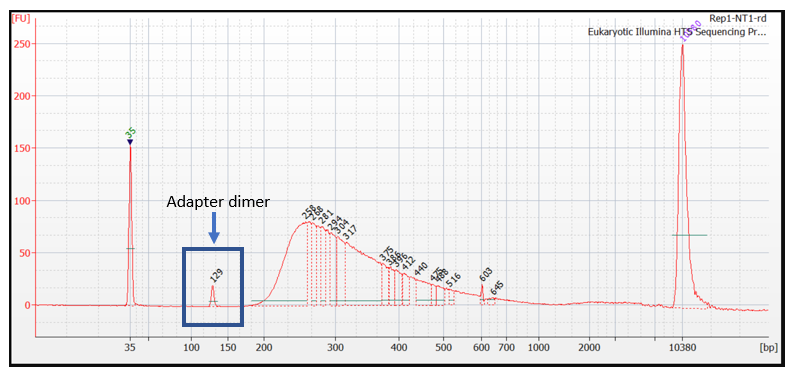
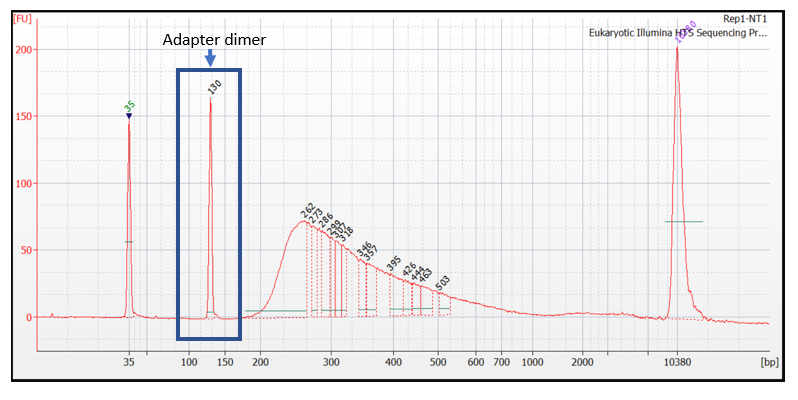
Figure 4. Bioanalyzer traces showing an adapter dimer peak between 120 bp and 150 bp.
- Diagnosing short inserts in a sequencing run through Q30 and %base composition
The effect of short inserts is reflected in the run metrics. Run metrics can be reviewed with Sequencing Analysis Viewer (SAV) software or BaseSpace Sequence Hub. Run statistics, particularly the Q30 scores, are helpful in diagnosing the presence of short inserts in a sequencing run. A rapid drop in the % > Q30, seen in insert reads, is indicative of short inserts.
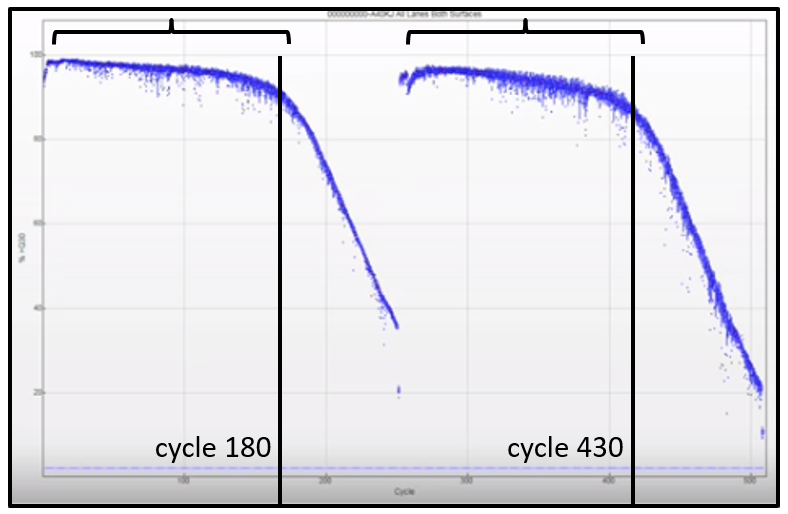
Figure 5. Drop in Q30 during a run. A sharp drop in the Q30 percentage at cycle 180 in both Read 1 and equivalent Read 2 indicates short inserts present in the DNA library.
When sequencing continues through the full DNA insert, proceeds through the adapter, and runs into the flow cell, the percent base profile will also change. This results in an A overcall on 4-channel instruments (MiSeq and HiSeq 2500) or a G overcall on 2-channel instruments (NextSeq550 and NovaSeq6000).
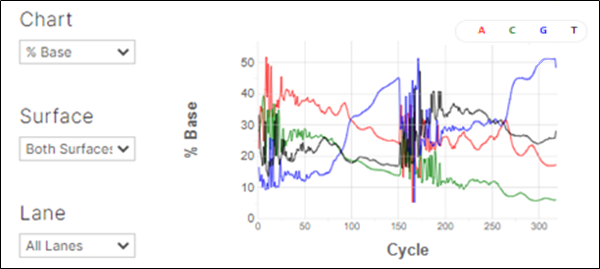
Figure 6. Percentage base composition. On 2-channel chemistry instruments such as the NextSeq or NovaSeq, the G-channel is the dark channel. If there is no base call, the software will assign a “G” read. Therefore, an increase in the G-channel in a 2-channel chemistry instrument can indicate the presence of short fragments.
- Confirmation of short inserts in data.
To confirm short inserts, the Adaptertrimming.txt file in the alignment output folder can be used. The output folder for the MiSeq is found here: MiSeq Analysis\{run folder}\Data\Intensities\Basecalls\Alignments\adaptertrimming.txt.
To determine the distribution of full length reads vs. trimmed reads, open the adaptertimming.txt file in Excel and plot the insert lengths of the library following adapter trimming. Then add up the bins of read length, and plot the results.
FastQC is also used to identify adapter content. The adapter section shows the adapter content as the read progresses.
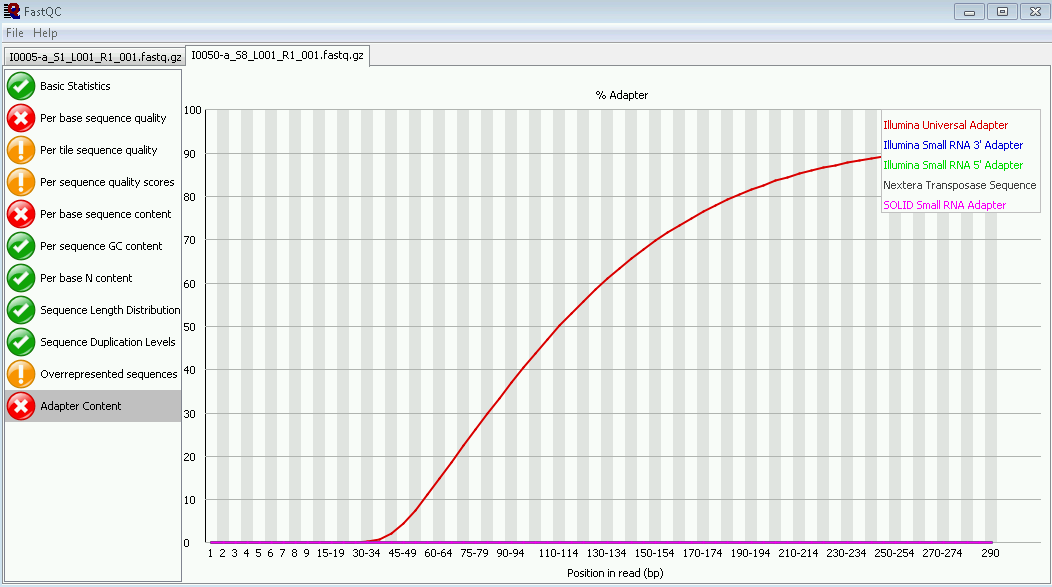
Figure 7. FastQC analysis. FastQC analyzes the sequencing run data for any adapter sequence.
Run data containing short inserts can still be used for analysis. Adapter sequences can be trimmed and removed from the sequencing read data, then the data can be further analyzed.







